Efficient all-electron hybrid density functionals for atomistic simulations beyond 10 000 atoms
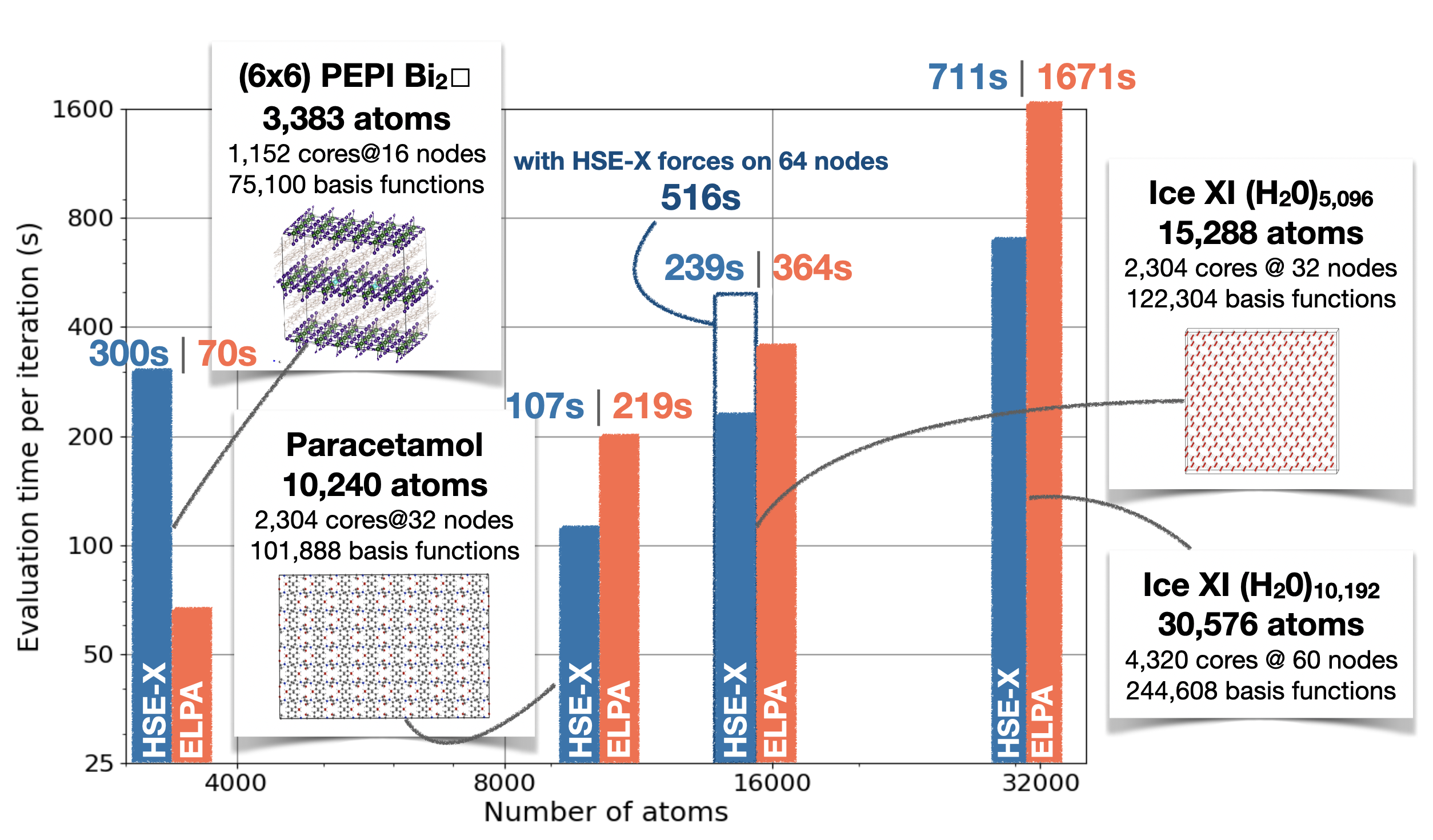
A highly optimized implementation of hybrid density functional approximations (DFAs) in the all-electron code FHI-aims has been developed, dramatically improving performance and scalability for both non-periodic and periodic systems. This collaborative effort extends the reach of hybrid DFAs to simulations of over ten thousand atoms, opening new possibilities for large-scale electronic structure calculations across diverse chemical systems, exemplified for perovskites, organic crystals, and complex ice structures.
read more